

Remember a few years year ago when your friends were pouring buckets of ice-cold water over the heads in the name of science? In total, a whopping $220 million was raised from the challenge. help anywhere from 12,000 to 15,000 people in US who reportedly live with ALS,
But did you know that sickle cell anemia (SCD) is the single most common life-threatening genetic disease in the United States? I sure didn’t. 
Over 100,000 Americans suffer from sickle cell disease and it is a torturous, painful life to live. Yet, the funding and publicity of sickle cell disease lags drastically far behind that of virtually every other genetic illness.
People with sickle cell disease have a lower-than-normal number of red blood cells because sickle cells don’t live as long as normal cells after they leave the bone marrow. Sickle cells usually die after about 10 to 20 days, compared to normal red blood cells, which live an average of 120 days. The bone marrow can’t make new red blood cells fast enough to replace all the dying ones. This causes anemia, low blood count that results in pain, fatigue, shortness of breath, and related symptoms
Sickle cell disease is inherited. Hemoglobin, the oxygen-carrying protein that is responsible for the red color of blood, is made of iron-containing heme and proteins called globins. People who have the disease inherit two copies of the globin gene that causes sickle cell disease. Those who live with the condition inherit a copy of the gene from each parent.
Contrary to popular belief, sickle cell disease is not a “black disease”. 
Sickle cell disease is most common in people whose families come from, or have ancestors from, sub-Saharan Africa, South or Central America (especially Panama), Caribbean islands, Mediterranean countries (such as Turkey, Greece, and Italy), India, and Saudi Arabia.
However, Sickle cell disease occurs in approximately one out of every 500 African American births and one out of every 36,000 Hispanic American births. About 2.5 million people in the United States have sickle cell trait, which occurs when a person inherits one copy of the globin gene.
Research has helped patients live longer. In the 1970s, life expectancy for individuals with sickle cell disease was about 14 years. Today, many individuals live into their 40s and longer.
There is no “moonshot” for sickle cell. There are no “ice bucket challenges. Fewer than a dozen US labs are working all-out on sickle cell disease, a number that has stayed constant for years. 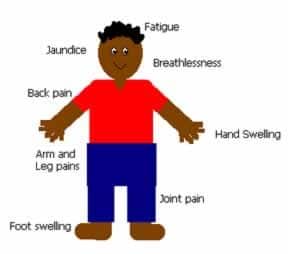
This is a disease that doesn’t get the attention or the funding it should. There’s a stigma that comes from thinking it affects only African-Americans. People dismiss it by saying, “Oh, sickle cell patients just have pain”.
The mainstays of treatment are transfusions and a drug called hydroxyurea. Hydroxyurea (HU) is an old drug approved by the FDA as part of some types of chemotherapy in 1967. It also is now the only FDA-approved therapy to prevent pain crises in SCD.
HU was approved for adults with SCD in 1998, 30 years after its approval for chemotherapy. There has been no commercial interest in developing a pediatric formulation, despite the need. The only curative treatment for SCD is bone marrow transplantation, which comes with high risks.
Pain from sickle cell disease crises is often undertreated as well. In part, this likely reflects racial and gender biases. Additionally, especially because the ischemic changes can’t be seen (unlike gangrene, for example, which is obvious), health care workers likely underestimate the person’s pain. Too often, these patients are treated as drug seekers.
Studies have shown that patients with SCD presenting to a hospital ER for care, during a crisis, experience longer wait times than other groups, even after accounting for assigned triage level. The African-American race of SCD patients, and their status as having SCD itself, both contribute to longer wait times for these patients. Where I come from, this is called “Institutional Racism”.
Undertreatment is likely fueled, too, by the feds focusing increased attention put on physician prescribing practices of narcotics. Regulations make it very cumbersome for physicians to prescribe adequate pain medications for chronic diseases. Patients, in turn, are subjected to humiliating drug screens to make sure that they are taking the narcotics and not selling them.
The funding disparities for research on sickle cell compared to other pediatric diseases are huge. Cystic fibrosis, a disease that affects primarily Caucasians, occurs in only a third of the numbers affected by SCD, but received 3.5 times more NIH funding. Private funding from foundations was about 400 times higher for cystic fibrosis!
There were 115 federally funded comprehensive care centers dedicated to cystic fibrosis. There were 10 for sickle-cell disease according to the study, which appeared in the journal Pediatrics in 2005.
One of the most challenging barriers to overcome in patients with sickle cell disease is transitioning from pediatric to adult health care.
Older patients with the condition may experience frequent visits to the emergency room, receive poor follow-up after hospital discharge and have limited access to medications that boost survival and lessen physical pain.

Despite the reality that four times the number of people suffer from sickle cell disease as those with cystic fibrosis, it’s clear that one genetic illness is advocated for far more than the other. While saying that racism is the lone cause of this disparity is an oversimplification of the problem, it’s certainly the foundation of it.
“In honor and memory of Deona Clark”
